 病历摘要 病历摘要
患儿男,5岁,因尿检异常10个月入院。患儿于10个月前患“水痘”后出现茶色尿,尿中泡沫较多。于当地医院查尿常规示蛋白(3+),潜血(3+),红细胞满视野,24小时尿蛋白定量288 mg,血白蛋白正常。4个月来患儿茶色尿、泡沫尿同前,无颜面、双下肢、阴囊水肿,期间定期查尿常规,蛋白(2+)~(3+)。行皮肤活检未见异常,予氯沙坦钾1/3片口服,每日1次,半月后自行停用。
患儿个人史无异常,生长发育正常,智力发育正常。
家族史:母亲31岁,双下肢水肿4年,乏力、夜尿增多1年。诊断为慢性肾炎,肾功能衰竭。肾脏病理活检显示,肾间质高度纤维增生,多灶性炎性细胞浸润;肾小管灶性萎缩;免疫荧光提示IgM(3+),C3(3+),IgA、IgM、IgG、C4、Fn、C1q均为阴性。Ⅳ型胶原连续阳性。
体格检查:全身皮肤无皮疹,颜面、双眼睑无水肿。心、肺、腹未见异常,双肾区无叩痛,双下肢、阴囊不肿。神经系统查体未见阳性体征。
实验室检查:
血常规WBC 7.5×109/L,N 54.2%,L 37.8%,Hb 112 g/L, Plt 352×109/L。
尿常规:蛋白(2+)~(4+),潜血(2+)~(3+),镜检红细胞3~4个/HP。24小时尿蛋白定量945 mg/24小时。血生化:尿素氮3.1 mmol/L,肌酐31.0 μmol/L,电解质、肝功能、心肌酶、凝血功能均正常。听力测试正常。
分析讨论
患儿为学龄前男童,起病隐匿,病史10个月,临床主要表现为茶色尿、尿中泡沫多,尿常规提示蛋白(2+)~(3+),镜检红细胞8~10个/HP,24小时尿蛋白定量945 mg,计算值41.0 mg/(kg・24 h),虽未达到大量蛋白尿诊断标准,亦提示存在肾病水平蛋白尿。患儿存在阳性家族史,其母亲有血尿、蛋白尿病史,当地医院诊断为慢性肾炎、肾功能不全,肾脏病理提示局灶节段性肾小球病变。故应高度怀疑患儿为家族遗传性肾脏疾病的可能,但须与以下几种疾病相鉴别。
1. 家族性局灶节段性肾小球硬化(FFSGS) 该病累及部分肾小球(<50%)有部分毛细血管袢的硬化性改变,小儿FFSGS平均起病年龄为6岁,男多于女,临床可仅表现为蛋白尿,或蛋白尿和血尿。54%~80%的FFSGS患儿伴有血尿,多为镜下血尿,伴有高血压、肾功能不全者分别为28%和20%。本病存在α肌动蛋白4(常染色体显性遗传)、Podocin突变(常染色体隐性遗传)及线粒体细胞病三种病因,确诊依靠病理检查。
2. Alport综合征(AS) 该病是一种肾小球基底膜(GBM)异常的遗传性肾脏病,临床上主要表现为血尿、感音神经性耳聋和进行性肾功能减退,其GBM超微结构呈特征性弥漫性的厚薄不均、分层、断裂。电镜显示GBM弥漫变薄,需肾脏病理检查以明确诊断。
3. 薄基底膜肾病(TBMD) 该病临床主要表现为镜下或肉眼血尿,不伴有肾功能障碍,无耳聋及眼部病变,确诊依靠病理检查。
4. IgA肾病 亦以血尿为主要表现,有一定家族倾向,因而需与之鉴别。
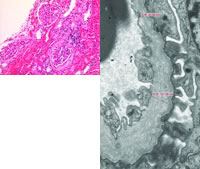
入院第6天完善肾脏病理检查:光镜(见图1)可见肾小球大致正常,一个包氏囊纤维化,未见嗜复红蛋白沉积,肾小管上皮细胞颗粒变性,间质可见淋巴细胞浸润,小血管未见异常,IgA、IgG、IgM,C3、C4,F,C1q均为阴性。
电镜(见图2)见肾小球毛细血管上皮细胞高度肿胀,足突阶段性融合,片状消失,毛细血管基底膜厚度不均,最窄处44.84 nm,最宽处121.36 nm,可见典型节段性基底膜撕裂分层及节段性基底膜菲薄,未见基底膜电子致密物沉积,内膜细胞肿胀,部分阻塞毛细血管。结合病史考虑病理检查符合AS诊断。
AS诊断标准
1996年Gregory等提出诊断AS的10条标准:(1) 有肾炎家族史,或先证者的一级亲属或女方的男性亲属中有不明原因的血尿。(2) 持续性血尿,无其他遗传性肾脏病的证据(如薄基底膜肾病、多囊肾或IgA肾病)。(3) 双侧2000~8000 Hz范围感音神经性耳聋,耳聋呈进行性,婴儿早期没有,但多于30岁前出现。(4) COL4A (3、4或5)基因突变。(5) 免疫荧光学检查显示,肾小球和皮肤基底膜完全或部分不表达Alport抗原决定簇。(6) GBM 的超微结构显示广泛异常,尤其是增厚、变薄和分裂。(7) 眼部病变,包括前圆锥形晶状体、后囊下白内障和视网膜斑点等。(8) 先证者或至少2例家系成员逐渐发展至终末期肾病(ESRD)。(9) 巨血小板减少症或白细胞包涵体。(10) 食管和女性生殖道的弥漫性平滑肌瘤。
若诊断AS家系,直系家庭成员必须符合4条标准(并不是同一例必须具备所有4条标准),当考虑旁系成员或仅表现为不明原因血尿、终末期肾病或听力障碍的极个别个体时应十分慎重;若判断AS家系中家庭成员是否受累时,如果该个体符合相应遗传型,且符合标准2~10中的1项,可作拟诊,符合2项便可诊断;对于无家族史患者的诊断。至少应符合4项指标。
该患儿肾脏病理和电镜结果均提示有典型AS改变,结合患儿家族史及临床表现,诊断AS尚存在以下疑点:
AS存在三种遗传方式,最常见的遗传型是X连锁显性遗传(XD),现已证实为编码基底膜Ⅳ型胶原α5链基因―COL4A5的突变所致。
患儿属于哪种遗传方式呢?
1. XD:此种遗传方式最常见,约占AS中的80%,AS基因携带者临床症状不显著,可仅表现为血尿,不伴有肾功能障碍,Ⅳ型胶原纤维检测可呈阳性表现。本例患儿及其母亲为发病者,且患儿母亲肾脏组织Ⅳ型胶原染色阳性,应首先考虑 XD遗传方式。但是患儿母亲病情重,31岁时便发生肾功能衰竭,诊断慢性肾脏病4期,与上述AS基因携带者临床表现不符。
2. 常染色体显性或隐性遗传(AR):约占AS中的10%~15%,这种遗传方式以COL4A3基因突变居多,由于致病基因在常染色体上,故遗传与性别无关,患者病情轻重也与性别无关。这就可以解释患儿母亲为什么病情重。但此种遗传方式发病率很低,且需要其父亲也为病变基因携带者才能解释患儿发病。
3. 基因突变:患儿肾脏活检免疫荧光染色均为阴性,而患儿母亲则为IgM3+,C33+,虽然在AS患者中可以出现IgM、C3阳性情况,但呈现3+阳性表现者鲜有报道,那么是否可以推测患儿母亲并不是AS基因携带者,患儿诊断AS与其母发生肾衰无关。以上推测需行基因分析以进一步协助诊断。
患儿皮肤组织、患儿母亲肾脏组织的Ⅳ型胶原染色阳性。有资料表明部分XD型男性GBM和皮肤基底膜上,α5染色阳性或弱阳性,其原因可能与基因突变特点有关。即免疫荧光显示α5、α4、α3阳性并不能除外AS诊断,可能是因为患儿α5链基因突变位点不同造成,某些基因突变位置可能对Ⅳ型胶原分子空间构象影响不大,抗原性仍然存在。另外Ⅳ型胶原分子的三股螺旋结构的形成开始于NC1区,发生了突变的α5可能没有影响在基底膜上形成Ⅳ型胶原分子三股螺旋结构的起始部分,且保留抗原性。故α5链显示正常,尚不能除外诊断。
总之AS是一种遗传性疾病,对于AS的诊断还是要结合临床表现、家系调查、电镜和Ⅳ型胶原检测进行综合诊断,对于诊断困难的病例需基因分析予以最终确诊,这样不仅可以确定突变基因特征、遗传方式,同时可以进行遗传、生育咨询。
图1(光镜HE×20),可见一个包氏囊纤维化,肾小管上皮细胞颗粒变性,间质可见淋巴细胞浸润
图2电镜(×15000) 可见足突阶段性融合,片状消失,毛细血管基底膜厚度不均,有节段性基底膜撕裂分层及节段性基底膜菲薄,未见基底膜电子致密物沉积。
|

